Algorytmy uczenia maszynowego, zaliczane do szeroko rozumianej „https://www.focus.pl/artykul/sztuczna-inteligencja-sprawniejsza-od-radiologow-w-diagnostyce-raka-piersi”, opracowane przez firmę DeepMind – spółkę-córkę Google’a – przewidziały trafnie strukturę 350 tysięcy białek. W tym większości białek organizmu człowieka. Odkrycie to może przynieść rewolucję w medycynie.
Zrozumienie struktury białek jest kluczowe m.in. dla medycyny
Wszystkie organizmy żywe składają się w dużej mierze z białek. Odgrywają one kluczową rolę dla życia. Poznanie budowy białek było więc od wielu dekad celem biochemików. Sekwencję aminokwasów – czyli ogniw, które tworzą białkowe łańcuchy, potrafimy określać dziś dokładnie i szybko.
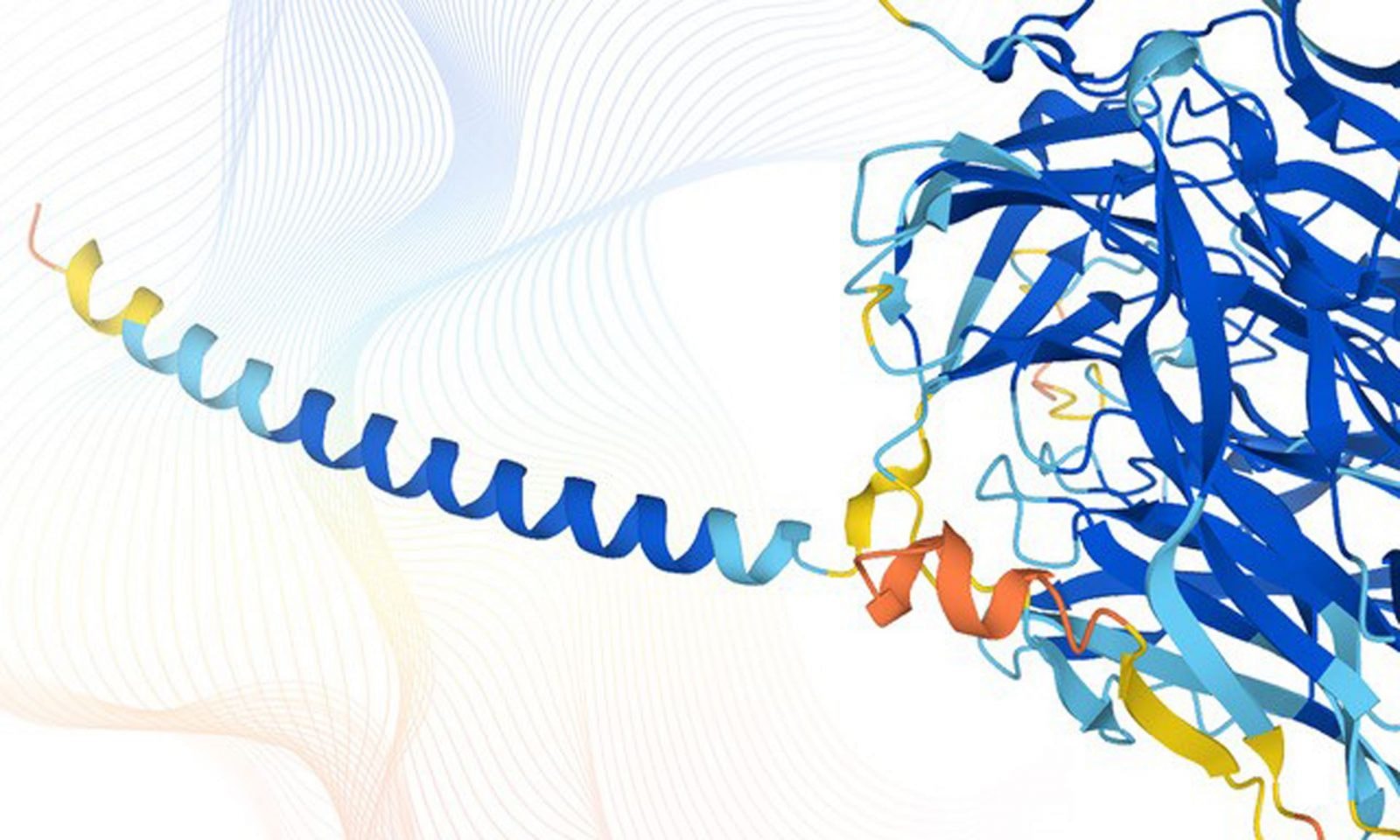
Białkowe łańcuchy są jednak poskręcane w trzech wymiarach w skomplikowane węzły. A właściwości białek zależą właśnie w dużej mierze od kształtu ich węzłów. Bada się je metodami eksperymentalnymi. To żmudna, czasochłonna praca, a metoda jest do tego kosztowna. Wymaga uzyskania czystego https://www.focus.pl/artykul/trening-w-pigulce-naukowcy-odkryli-bialko-ktore-imituje-korzystne-efekty-wysilku-fizycznego, jego krystalizacji i badania kryształu za pomocą prześwietlania promieniami rentgena. W przypadku niektórych białek w ogóle nie jest to możliwe.
DeepMind i badacze z Europejskiego Laboratorium Biologii Molekularnej w ubiegłym tygodniu ogłosili przełom na łamach prestiżowego czasopisma „Nature”. Za pomocą algorytmu uczenia maszynowego o nazwie AlphaFold udało się im rozszyfrować strukturę ponad 350 tysięcy białek. W tym około 20 tysięcy, czyli większość, białek organizmu człowieka.
Badanie struktury białek za pomocą komputerów będzie szybsze i tańsze
W miarę rozwoju technologii komputerowych powstała nadzieja, że zamiast badać kształt białka, można będzie przeprowadzić komputerowe symulacje, jak dana sekwencja aminokwasów się zwinie. Sposób działał jednak tylko w przypadku krótkich https://www.focus.pl/artykul/bialko-z-ludzkich-wlosow-pomoze-w-leczeniu-czerniaka, gdzie kombinacji aminokwasów jest niewiele.
W przypadku długich łańcuchów białkowych nawet superkomputery nie dawały sobie rady. A właściwie nawet nie było sensu ich uruchamiać. Dla przykładu, gdyby każdy aminokwas mógł tylko występować w dwóch możliwych pozycjach, w białku złożonym ze stu aminokwasów liczba możliwych ułożeń wynosiłaby aż 2 do potęgi 100. To około 10 do potęgi 30, czyli trylion trylionów. Liczenie tego krok po kroku zajmowałoby długie lata.
Rozwiązaniem okazało się wykorzystanie algorytmów uczenia maszynowego. Omija to konieczność wyliczania struktury białka krok po kroku. Taki algorytm nic nie wylicza. Na podstawie przykładów uczy się, że dane sekwencje aminokwasów zwijają się tak, zaś inne inaczej. Na podstawie krótszych sekwencji przewiduje zaś dłuższe.
Przewidywanie struktur białek za pomocą algorytmów uczenia maszynowego rozwijano od ponad dekady. Pierwsze próby były niezbyt udane. Przełomowy okazał się dopiero algorytm AlphaFold przedstawiony przez DeepMind w 2019 roku. Przewidział trafnie strukturę większości białek, choć wówczas było ich zaledwie kilkanaście.
Sztuczna inteligencja przewiduje strukturę białek
Potem autorzy algorytmu zamilkli. Dość zagadkowo, lecz dziś wiemy, że pracowali nad rozszyfrowaniem przestrzennych struktur setek tysięcy białek. W „Nature” ogłosili, że AlphaFold rozpracował większość białek ludzkich, bo aż 58 proc. (z tego 35,7 proc udało się wymodelować z „dużą pewnością”).
Dla porównania, dziesięciolecia prac biochemików w laboratoriach pozwoliły na poznanie struktury zaledwie 17 proc. białek. Prof. John McGeehan, biolog z Uniwersytetu w Portsmouth komentuje dla BBC News, że to, co zajmowało do tej pory miesiące prac w laboratorium, algorytm wykonuje w kilka minut.
– Gdy przesłaliśmy siedem sekwencji aminokwasów do zespołu DeepMind, struktury dwóch białek już znaliśmy z badań eksperymentalnych. Gdy wyniki do nas wróciły, mogliśmy je więc potwierdzić. Szczerze mówiąc była to chwila, w której włosy stanęły mi dęba. Struktury przewidziane przez algorytm były identyczne – dodaje naukowiec.
Poznanie struktury białek to przełom w medycynie
Prof. Edith Heard z Europejskiego Laboratorium Biologii Molekularnej mówi, że ten przełom zupełnie zmieni nasze pojmowanie mechanizmów życia. Zastosowanie tej metody zaś ogranicza tylko ludzki umysł.
Znając dokładny kształt białka można bowiem łatwiej opracować lek, który się do białka dopasuje. Pozwoli to na opracowywanie lepszych leków, także na choroby, które dotychczas były nieuleczalne. Do tej pory leków także szukano nieco po omacku, metodą prób i błędów. Jeśli znamy jednak kształt białka, które chcemy np. zablokować, lek można dopasować będzie do niego niczym klucz do zamka.
Inne zastosowania to opracowanie enzymów, które mogą rozkładać tworzyw sztuczne, nad czym zespół prof. McGeehana właśnie pracuje. Przyspieszy to ich prace, jak mówi, o wiele lat. I da szanse na uporanie się z coraz większym problemem, jaki stwarzają plastikowe śmieci.
Struktury białek opisywane przez Europejskie Laboratorium Biologii Molekularnej przy pomocy AlphaFold będą dostępne dla naukowców na całym świecie. Docelowo DeepMind chciałby, żeby w bazie danych znalazły się wszystkie białka znane naukowcom. W sumie jest ich aż 100 milionów.
DeepMind przyśpieszył prace pod wpływem konkurencji ze strony niezależnych biologów
Trzeba zaznaczyć, ze za tym przełomem stoi algorytm opracowany przez spółkę należącą do https://www.focus.pl/artykul/sztuczna-inteligencja-na-celowniku-google’a, który kupił DeepMind w 2014 roku. Nie należy się spodziewać, że każdy badacz będzie mógł wrzucić do algorytmu AlphaFold znaną sekwencje aminokwasów i za darmo otrzymać jej przestrzenną strukturę.
Odkrycie ogłoszono zresztą w pośpiechu właśnie dlatego, że spółce DeepMind zaczęli po piętach deptać naukowcy. Zniecierpliwiło ich to, że od grudnia 2020 roku, kiedy firma doniosła o znakomitych wynikach algorytmu AlphaGo, nie podała żadnych szczegółów.
Część biologów, zajmująca się badaniem struktur białek zaczęła się zastanawiać, czy nie powinna zmienić zawodu. Wpadli jednak na inny pomysł – stworzenia własnego algorytmu. W ciągu pół roku udało im się stworzyć od podstaw https://www.focus.pl/artykul/decyzje-z-czarnej-skrzynki, który nazwali RoseTTAfold .
Opublikowali swoją pracę w „Science” tydzień przed doniesieniem DeepMind. Przyznają, że komercyjny algorytm jest nieco lepszy. Jednak ich oprogramowanie jest jednak dostępne za darmo na serwerze utrzymywanym przez badaczy.